Om sicklecellanemi
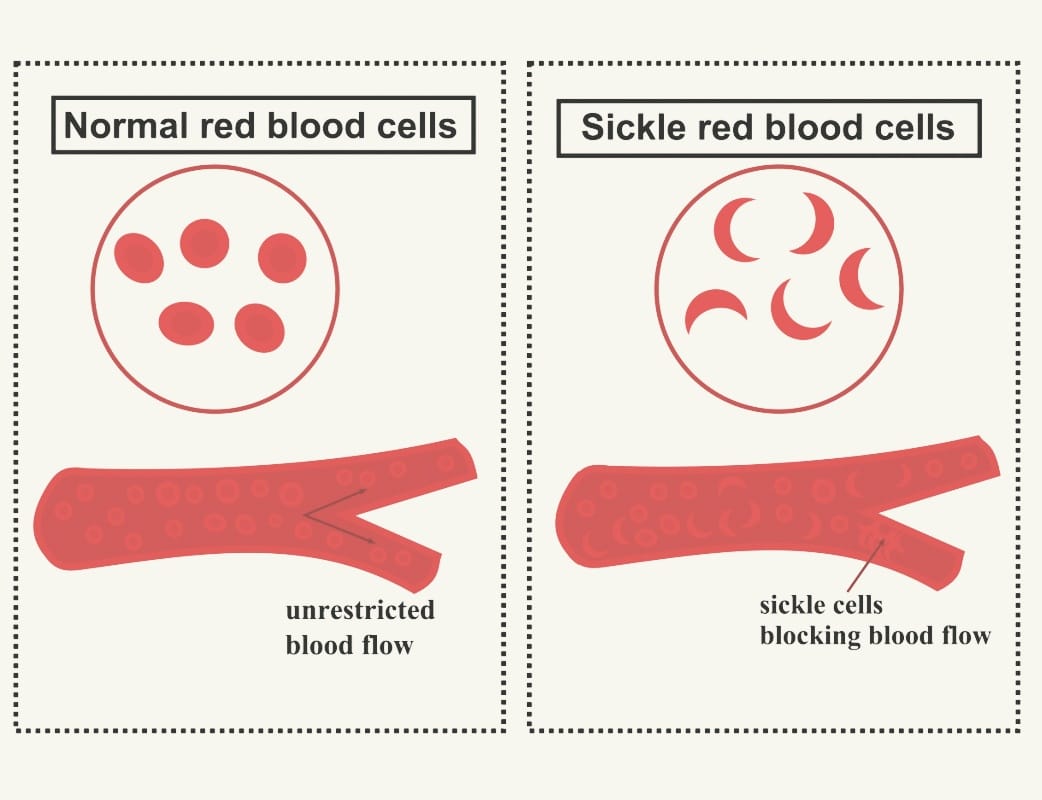
18 augusti 2025Sicklecellanemi är en ärftlig blodsjukdom som påverkar hemoglobinets struktur och funktion. Hemoglobinet är det syretransporterande proteinet i de röda blodkropparna, och vid sicklecellanemi är det förändrat på ett sådant sätt att cellerna kan anta en stel, halvmåneformad struktur, så kallade sickleceller. Dessa missformade celler fastnar lätt i små blodkärl, vilket kan orsaka blockeringar, smärta, syrebrist i vävnader och långsiktiga organskador. Tillståndet är kroniskt och varierar i svårighetsgrad, men obehandlat kan det ge allvarliga komplikationer och förkortad livslängd.
Sjukdomen beskrevs första gången 1910 av den amerikanske läkaren James Herrick, som observerade ovanliga, långsmala röda blodkroppar hos en patient med anemi. På 1940-talet identifierades orsaken som en förändring i hemoglobinmolekylen, och sicklecellanemi blev därmed den första sjukdomen som kopplades till en specifik mutation i en gen. Detta banade väg för förståelsen av molekylärgenetikens roll i ärftliga sjukdomar.
Sicklecellhemoglobin
Sicklecellhemoglobin (HbS) är en variant av det normala hemoglobinet (HbA) som orsakas av en specifik mutation i β-globingenen (HBB) på kromosom 11. Mutationen leder till ett aminosyrautbyte där glutamat ersätts med valin på position 6 i β-globinkedjan (β6 Glu→Val). Denna förändring gör att hemoglobinmolekylerna kan klumpa ihop sig när syretrycket sjunker, vilket leder till att de röda blodkropparna blir stela och antar en karakteristisk halvmåneform, så kallade sickleceller. Sickleformen gör att cellerna har svårare att passera genom små blodkärl, vilket ger upphov till blockeringar och vävnadsskada. Cellerna bryts även ned snabbare än normalt (livslängd 10–20 dagar jämfört med 120 dagar för friska celler). Resultatet är en kombination av blockeringar (vaso-ocklusion) och kronisk blodbrist (hemolys).
Genetik och nedärvning
Sicklecellanemi ärvs autosomalt recessivt vilket innebär att sjukdomen uppstår när en person har ärvt två förändrade kopior av β-globingenen, en från vardera föräldern. ”Autosomalt” betyder att genen sitter på en av de 22 icke-könskromosomerna, vilket gör att både män och kvinnor kan drabbas i samma utsträckning. ”Recessivt” betyder att en enda muterad gen inte räcker för att sjukdomen ska utvecklas, individen måste ha två förändrade gener (en homozygot uppsättning, HbSS).
Om en person däremot bara bär på en muterad gen och en normal gen (heterozygot, HbAS), får hen vanligtvis inte sjukdomen utan blir bärare, vilket kallas sicklecelltrait. Dessa bärare har sällan symtom, men kan föra anlaget vidare till sina barn. När två bärare får barn är risken 25% att barnet får sjukdomen, 50% att det blir bärare, och 25% att det är helt friskt utan anlag. Personer med sicklecelltrait är oftast symtomfria, men kan under vissa omständigheter som extrem fysisk ansträngning eller hypoxi, uppleva milda symtom. Kombinationer med andra hemoglobinvarianter, som HbC eller β-thalassemi som också är en ärftlig sjukdom, men där produktionen av β-globinkedjor är nedsatt eller saknas, kan ge mellansvåra former.
Ärftlighetstabell
Tabellen nedan visar hur olika kombinationer av föräldrarnas genvarianter för sicklecellhemoglobin kan resultera i olika möjliga genotyper hos barnet och den procentuella sannolikheten för varje utfall. Listan täcker de vanligaste kliniskt relevanta kombinationerna mellan HbA, HbS och HbC som leder till sicklecellsjukdom (HbSS, HbSC) eller anlagsbärarskap (HbAS). HbAC och HbCC ingår som C-relaterade tillstånd, men räknas inte som sicklecellsjukdom.
| Förälder 1 | Förälder 2 | Möjliga genotyper hos barnet | Sannolikhet |
|---|---|---|---|
| HbAA | HbAA | HbAA | 100% |
| HbAA | HbAS | HbAA, HbAS | 50%, 50% |
| HbAA | HbSS | HbAS | 100% |
| HbAA | HbAC | HbAA, HbAC | 50%, 50% |
| HbAA | HbSC | HbAS, HbAC | 50%, 50% |
| HbAA | HbCC | HbAC | 100% |
| HbAS | HbAS | HbAA, HbAS, HbSS | 25%, 50%, 25% |
| HbAS | HbSS | HbAS, HbSS | 50%, 50% |
| HbAS | HbAC | HbAA, HbAS, HbAC, HbSC | 25% vardera |
| HbAS | HbSC | HbAS, HbAC, HbSS, HbSC | 25% vardera |
| HbAS | HbCC | HbAC, HbSC | 50%, 50% |
| HbSS | HbSS | HbSS | 100% |
| HbSS | HbAC | HbAS, HbSC | 50%, 50% |
| HbSS | HbSC | HbSS, HbSC | 50%, 50% |
| HbSS | HbCC | HbSC | 100% |
| HbAC | HbAC | HbAA, HbAC, HbCC | 25%, 50%, 25% |
| HbAC | HbSC | HbAS, HbAC, HbSC, HbCC | 25% vardera |
| HbAC | HbCC | HbAC, HbCC | 50%, 50% |
| HbSC | HbSC | HbSS, HbSC, HbCC | 25%, 50%, 25% |
| HbSC | HbCC | HbSC, HbCC | 50%, 50% |
| HbCC | HbCC | HbCC | 100% |
HbAA – Normalt hemoglobin
Hos personer med genuppsättningen HbAA finns två normala kopior av β-globingenen. Det innebär att hemoglobinet är normalt (HbA) och att individen inte har någon ökad risk för sicklecellrelaterade symtom. Dessa personer kan endast föra vidare normala gener och om båda föräldrar har HbAA kan barnet inte få sicklecellsanlag.
Hos vuxna med denna normala genuppsättning utgör hemoglobin A (HbA) vanligen cirka 95–98% av allt hemoglobin. Den återstående andelen består främst av hemoglobin A₂ (HbA₂) på ungefär 2–3% samt mycket små nivåer av fosterhemoglobin (HbF), oftast mindre än 1%. Förskjutningar i denna fördelning kan tyda på ärftliga hemoglobinförändringar, olika blodsjukdomar eller effekter efter nyligen givna blodtransfusioner.
HbAS – Sicklecelltrait / Anlagsbärare
Genuppsättningen HbAS, även kallad sicklecelltrait eller anlagsbärare, innebär att en normal gen (HbAA) kombineras med en gen med sicklecellsförändring (HbAS).
Hos personer med sicklecelltrait (HbAS) består blodet vanligtvis av cirka 55–60% normalt hemoglobin A (HbA) och 35–45% sicklecellhemoglobin (HbS). Därtill finns en liten andel HbA2 på omkring 2–3% och oftast mindre än 1% HbF. Eftersom HbA dominerar över HbS är personer med HbAS i regel symtomfria, även om vissa kan få problem vid extrem fysisk belastning eller syrebrist. Om båda föräldrar har HbAS finns 25% risk att barnet får sicklecellanemi (HbSS).
HbSS – Sicklecellanemi
Vid HbSS finns två gener med sicklecellsförändring, vilket leder till sjukdomen sicklecellanemi. Detta ger kronisk blodbrist, återkommande smärtkriser, ökad infektionsbenägenhet och risk för organskador. Sjukdomen kräver livslång medicinsk uppföljning och behandling. En person med HbSS för alltid vidare minst en HbS-gen till sina barn.
Hos personer med sicklecellanemi (HbSS) består en stor del av hemoglobinet av sicklecellhemoglobin (HbS), ofta över 90–95%. En mindre andel kan utgöras av HbF (fosterhemoglobin), vars nivå varierar men ibland är förhöjd och kan mildra sjukdomsbilden. Hemoglobin A (HbA) saknas helt vid HbSS.
HbAC – Hemoglobin C-anlag
Hos individer med HbAC finns en normal gen (HbA) och en gen för hemoglobin C (HbC). Dessa personer är oftast symtomfria, men kan ibland ha lätt blodbrist. Om en person med HbAC får barn med någon som har HbS finns en möjlighet att barnet får HbSC-sjukdom.
Hos personer med HbAC brukar hemoglobinfördelningen se ut så att hemoglobin A (HbA) utgör omkring 50–60% av det totala hemoglobinet, medan hemoglobin C (HbC) står för ungefär 30–40%. Andelen hemoglobin A2 (HbA₂) ligger vanligen kvar på normala nivåer kring 2–3% och hemoglobin F (HbF) utgör mindre än 1 procent. Fördelningen kan variera något mellan individer, men mönstret kännetecknas av att både HbA och HbC finns i betydande mängd.
HbSC – Kombinerad sicklecellsjukdom
HbSC innebär en kombination av en HbS-gen och en HbC-gen. Detta är en form av sicklecellsjukdom som ofta är mildare än HbSS, men som ändå kan orsaka smärtkriser, blodbrist och organskador.
Vid HbSC saknas normalt hemoglobin A (HbA) helt. Hemoglobinfördelningen består istället huvudsakligen av HbS och HbC i ungefär lika delar, vanligen omkring 45–50% vardera. HbA₂ ligger ofta kvar på normala nivåer runt 2–3%, och små mängder HbF (<1%) kan ibland förekomma. Denna balans mellan HbS och HbC bidrar till sjukdomsbilden, som ofta är mildare än vid homozygot HbSS, men ändå kan ge återkommande symtom.
HbCC – Hemoglobin C-sjukdom
HbCC betyder att båda β-globingenerna kodar för hemoglobin C. Tillståndet ger vanligen mild hemolytisk anemi, ibland med lätt gulsot och förstorad mjälte, men saknar de typiska smärtkriserna som ses vid HbSS och HbSC. De flesta lever utan större begränsningar, men kan behöva regelbundna kontroller. Vid nedärvning kan en HbCC-förälder bara föra vidare HbC-genen, vilket får betydelse om den andra föräldern bär på HbS (risk för HbSC hos barnet).
Vid HbCC saknas normalt HbA helt. Hemoglobinet består nästan uteslutande av HbC, vanligen omkring 90–98%. HbA₂ ligger ofta normalt eller lätt förhöjt (cirka 2–4%), och HbF är lågt men kan vara lätt förhöjt (oftast <5%, hos barn ibland något högre). Andelar varierar med ålder och eventuella transfusioner och påvisat HbA talar i regel för nyligen given blodtransfusion.
Andra kombinationer
Utöver A/S/C-kombinationer kan andra hemoglobinvarianter förekomma tillsammans med HbS och med β-thalassemi. HbS i kombination med HbD-Punjab, HbE eller HbO-Arab kan ge HbSD, HbSE respektive HbSO, och HbS tillsammans med β-thalassemi ger S/β-thal, där sjukdomsbilden oftast är mer uttalad vid β⁰ än vid β⁺.
HbSD – Sickle-Hemoglobin D-sjukdom
HbSD innebär att en HbS-gen kombineras med en HbD-gen, oftast varianten HbD-Punjab. Sjukdomsbilden liknar i många fall sicklecellanemi, med smärtkriser och risk för komplikationer, även om den kan vara något mildare. Tillståndet är vanligare i delar av Indien, Pakistan och Medelhavsområdet.
HbSE – Sickle-Hemoglobin E-sjukdom
HbSE är en kombination av en HbS-gen och en HbE-gen. Tillståndet ger oftast en mildare sjukdomsbild än HbSS, men det finns rapporterade fall med smärtkriser och anemi. HbE är vanlig i Sydostasien, och kombinationen ses oftast hos personer med ursprung från denna region.
HbSO – Sickle-Hemoglobin O-Arab-sjukdom
HbSO innebär att en HbS-gen kombineras med en HbO-Arab-gen. Detta kan ge en sjukdomsbild som liknar sicklecellanemi, ibland med lika svåra symtom, inklusive smärtkriser, blodbrist och risk för organskador. HbO-Arab är vanligast i Mellanöstern, Nordafrika och vissa delar av Grekland.
S/β-thal – Sickle–β-thalassemi
S/β-thal innebär att en HbS-gen kombineras med en β-thalassemi-gen, antingen β⁰ (ingen β-kedjeproduktion) eller β⁺ (nedsatt produktion). Sjukdomsbilden påminner ofta om sicklecellanemi med smärtkriser, hemolytisk anemi och risk för organskador. Svårighetsgraden är vanligen högre vid S/β⁰ än vid S/β⁺, eftersom frånvaron av HbA vid β⁰ ger mer uttalad sickling. Tillståndet ses främst i områden där β-thalassemi är vanlig, som delar av Medelhavet, Mellanöstern och Sydasien.
Klinisk bild och komplikationer
Sicklecellanemi ger återkommande smärtkriser orsakade av att sickleceller fastnar i små blodkärl och hindrar blodflödet. Detta leder till syrebrist i vävnader och kan orsaka allvarlig smärta, särskilt i skelett och buk. Kronisk hemolytisk anemi är vanligt, vilket ger symtom som trötthet, blekhet och gulsot. Sjukdomen ökar risken för infektioner, stroke, akut bröstsyndrom, njurpåverkan och gradvis förlust av mjältfunktion. Hos barn kan man se försenad tillväxt och pubertetsutveckling.
Akuta komplikationer och livshotande episoder
Återkommande smärtkriser
Ett av de mest framträdande akuta symtomen vid sicklecellsjukdom är återkommande smärtattacker. Smärtan uppstår när de missformade blodkropparna fastnar i små kärl och blockerar blodflödet, ett tillstånd som kallas vaso-ocklusiv kris. Detta leder till syrebrist i vävnaden, särskilt i benmärgen, och orsakar ofta svår skelettsmärta. Smärtan kan dock uppträda i andra delar av kroppen och feltolkas ibland som andra sjukdomstillstånd. Utlösande faktorer kan vara kyla, infektioner eller vätskebrist, men i många fall saknas en tydlig orsak.
Smärtskov i skelettet
Plötsliga och intensiva smärtor i benstommen är det vanligaste akuta symtomet. Dessa orsakas av att blodflödet blockeras av sickleceller och syrebrist uppstår i benmärgen. Krisen kan utlösas av infektion, kyla eller uttorkning men kommer ofta utan tydlig anledning.
Akuta bröstbesvär
Akut bröstsyndrom innebär bröstsmärta, feber och andningssvårigheter. Tillståndet kan snabbt bli livshotande och ses ofta som lunginfarkt eller infektion i kombination med sickling i lungkärlen.
Infektionskänslighet
Patienter med sjukdomen har en ökad mottaglighet för bakteriella infektioner, exempelvis blodförgiftning, lunginflammation eller mer ovanliga infektioner i bukhinna och skelett.
Tillfälligt bortfall av blodbildning
Vid vissa virusinfektioner kan benmärgens produktion av blodkroppar stanna av, vilket leder till kraftig blodbrist på kort tid. Symtomen kan vara trötthet, blekhet, hjärtklappning och andningssvårigheter. Tillståndet är oftast övergående.
Stroke hos barn och vuxna
Förträngningar i hjärnans blodkärl kan orsaka stroke. Hos yngre rör det sig ofta om blodproppar, medan blödningar och TIA (Transitorisk Ischemisk Attack som innebär en tillfällig syrebrist i hjärnan) blir vanligare senare i livet. Neurologiska symtom som förlamning, svaghet eller plötslig huvudvärk kan förekomma.
Blodansamling i mjälten
Hos små barn kan stora mängder blod fastna i mjälten. Detta leder till akut blodbrist, cirkulationssvikt och snabbt livshotande tillstånd. Tidiga symtom är trötthet, buksmärta, blekhet och snabb puls.
Priapism – ihållande erektion
Män och pojkar kan drabbas av smärtsamma, långvariga erektioner, ofta nattetid. Orsaken är inte alltid känd, men tillståndet är återkommande och kan vara mycket plågsamt.
Långvariga följder av sicklecellsjukdom
Försenad kroppslig utveckling
Barn med sicklecellsjukdom har ofta en långsammare tillväxtkurva och når puberteten senare än jämnåriga. De flesta växer dock ikapp i vuxen ålder.
Nedsatt njurfunktion
Njurar som utsätts för återkommande syrebrist förlorar gradvis sin förmåga att koncentrera urinen. Detta leder till ökad urinproduktion, ständig törst och risk för vätskebrist. På längre sikt kan njurskador utvecklas och i vissa fall leda till njursvikt.
Synproblem kopplade till näthinnan
Förändringar i näthinnans blodkärl (retinopati) kan göra att nya, sköra blodkärl bildas. Dessa kan brista och orsaka blödningar som påverkar synen.
Sexuella komplikationer
Återkommande episoder av priapism hos män kan med tiden resultera i nedsatt erektionsförmåga.
Kognitiva och neurologiska följder
Upprepade proppar i hjärnans blodkärl förekommer, särskilt hos barn, och kan påverka minne, inlärning och andra kognitiva förmågor.
Bensjukdomar och vävnadsdöd
Otillräcklig blodtillförsel till lederna kan ge nekros i höft- eller axelleden, ofta i 25–30-årsåldern. Detta orsakar smärta och begränsad rörlighet.
Kroniska lungförändringar
Många utvecklar andningsproblem, bröstsmärtor och nedsatt kondition. På sikt kan även högt blodtryck i lungornas kärl uppstå.
Sår på underbenen
Svårläkta bensår, ofta kring anklarna, är vanliga i vuxen ålder. De är smärtsamma, återkommer lätt och kan leda till infektion.
Gallstenssjukdom
Ökad nedbrytning av röda blodkroppar ger ökad bilirubinproduktion och därmed en högre risk för gallsten. Förekomsten ökar med åldern och kan ge både smärta och bukrelaterade komplikationer.
Diagnostik
Diagnosen baseras ofta på blodstatus som visar normocytär anemi (normala, men för få röda blodkroppar), retikulocytos (ökad mängd omogna röda blodkroppar) och hemolys (för tidig nedbrytning av röda blodkroppar).
Analysen hemoglobinfraktioner, som utförs med metoder som högpresterande vätskekromatografi (HPLC) eller hemoglobinelektrofores, används för att påvisa förekomst av HbS samt för att mäta fördelningen mellan olika hemoglobiner, exempelvis HbA, HbA₂ och HbF. Vid homozygot sicklecellanemi (HbSS) saknas HbA helt. För att bekräfta diagnosen och identifiera kombinationer med andra varianter, som HbC eller β-thalassemi, kompletteras ofta utredningen med genetisk analys av HBB-genen.
Upptäckt av HbSS och HbAS
I många länder ingår sicklecelltest i nyföddhetsscreening för att snabbt kunna identifiera drabbade barn. I Sverige är testning vanligast vid utredning av anemi eller hos personer med ursprung från högendemiska områden.
HbSS (sicklecellanemi) upptäcks vanligen tidigt, antingen via nyföddhetsscreening i länder där det ingår eller genom att barnet blir sjukt när andelen HbF sjunker vid 4–6 månaders ålder (till exempel anemi, smärtepisoder, infektioner). HbAS (sicklecellanlag) ger däremot sällan symtom och hittas därför ofta först i vuxen ålder, till exempel vid utredning av anemi, inför graviditet/partnertestning, vid hälsokontroller (idrott/arbete), blodgivning eller familjeutredning. Undantaget är länder/regioner med systematisk bärarscreening, där kan HbAS identifieras redan i barndomen.
Behandling och framtidsutsikter
Behandlingen syftar till att förebygga och hantera komplikationer. Hydroxyurea (hydroxykarbamid) är ett läkemedel som hämmar enzymet ribonukleotidreduktas och därmed minskar DNA-syntesen. Inom sicklecellsjukdom används det för att öka andelen HbF (fosterhemoglobin), vilket gör att röda blodkroppar sicklar mindre. Effekten blir färre smärtkriser, lägre risk för akut bröstsyndrom och färre sjukhusinläggningar.
Regelbundna blodtransfusioner ges vid särskilda risker, till exempel hög risk för stroke. Benmärgstransplantation är i vissa fall botande, men används främst vid svåra former. Genterapier, inklusive CRISPR-baserade metoder, är under snabb utveckling och har godkänts i vissa länder.
Evolutionär aspekt och skydd mot malaria
Bärare av sicklecellanlaget (HbAS) har ett visst skydd mot svår malaria genom att deras röda blodkroppar till största delen är normala, men under syrebrist kan en andel anta sickleform. Detta gör att malariaparasiten Plasmodium falciparum får svårare att utvecklas eftersom infekterade celler bryts ner snabbare och rensas bort av immunförsvaret eller fastnar i mjälten innan parasiten fullbordat sin livscykel. Effekten minskar risken för allvarlig malaria, även om bäraren kan bli smittad. I områden där malaria länge varit utbrett har detta inneburit en evolutionär fördel, vilket bidragit till att HbS-genen bevarats i hög frekvens, trots att två kopior av genen (HbSS) leder till allvarlig sjukdom.
Personer med sicklecellanemi (HbSS) saknar malariaskyddet som bärare (HbAS) har eftersom sjukdomen orsakar kronisk blodbrist, skadad mjältfunktion och nedsatt immunförsvar, vilket gör malaria allvarligare. Trots detta är HbS-genen vanlig i malariaområden på grund av balanserande selektion, där bärarskap (en HbS-kopia) ger fördel i form av skydd mot svår malaria, medan två kopior leder till sjukdom.
Förekomst i Sverige och världen
Globalt föds omkring 300 000 barn varje år med sicklecellanemi, främst i Afrika söder om Sahara, Mellanöstern och delar av Indien. I Sverige är sjukdomen ovanlig i den inhemska befolkningen, men förekommer hos personer med ursprung i högendemiska områden. Antalet diagnostiserade fall har ökat på grund av migration och ökad medvetenhet. Specialistmottagningar finns vid flera universitetssjukhus.
Källor
Socialstyrelsen.se – Sicklecellsjukdom
Lakartidningen.se – ABC om sicklecellsjukdom
Agrenska.se – Information om sicklecellanemi (Ågrenska)
Blodcancerforbundet.se – Broschyr om sicklecellanemi
PHO.barnlakarforeningen.se – Riktlinjer för sicklecellanemi (Barnläkarföreningen)
Beställ blodprov för sicklecellanemi och β-thalassemi







